Maladie génétique rare
Le pari scientifique qui a sauvé lyana
La Presse

Cet écran a été partagé à partir de La Presse+
Édition du 30 avril 2019,
section ACTUALITÉS, écran 5


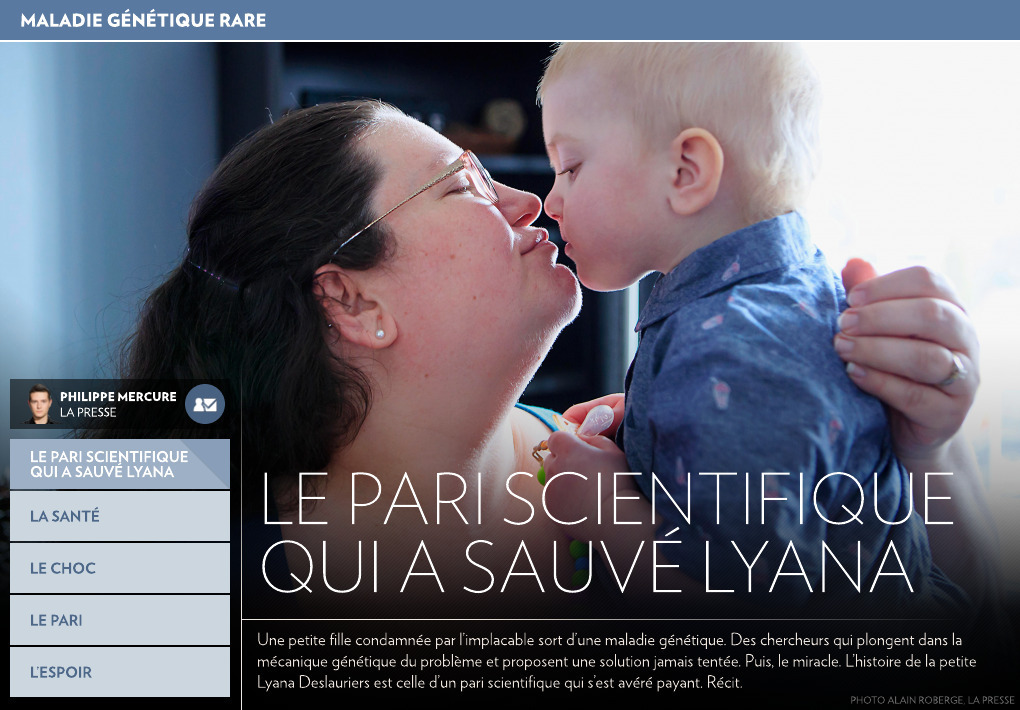
Lyana Deslauriers a une couleur préférée : le rouge. Quand la petite fille a fêté son deuxième anniversaire, le 21 novembre dernier, la maison familiale au grand complet est devenue écarlate.
« Les ballons rouges, les guirlandes rouges… Tout ce qui pouvait être rouge, on en a mis en masse », raconte son père Joël.
« Elle avait 20 % de chances de se rendre à 2 ans, alors les 2 ans, on les attendait en tabarouette. Et on les a fêtés en tabarouette aussi. Je pense que ça a duré un mois », ajoute Lynn Ménard, la mère de la petite.
Pendant que ses parents racontent son histoire, Lyana, aucunement intéressée, file sur le balcon jouer avec sa « tour d’eau ». Malgré la pluie et le froid de ce printemps qui tarde à s’installer, elle transvide de l’eau dans des seaux et rit en regardant les petites roues de plastique tourner au gré du courant. Lyana est la cinquième enfant d’une famille de six, qui est sur le point de s’agrandir encore avec la naissance prochaine d’un garçon. Son frère et ses trois grandes sœurs se relaient, parapluie à la main, pour la protéger pendant qu’elle joue.
Sa présence ici, dans cette chaleureuse maison de Saint-Charles-Borromée où l’action ne manque pas, est une sorte de miracle.
« Savoir que cette enfant a une vie normale et des activités normales… C’est incroyable », dit le Dr Gregor Andelfinger, chercheur et cardiologue pédiatre au CHU Sainte-Justine, qui a joué un rôle crucial dans cette étonnante entorse au destin.
Pour Lynn Ménard et Joël Deslauriers, les premiers doutes surgissent lors d’une échographie effectuée à la 32e semaine de grossesse.
Les tests suivants confirment les soupçons : le cœur présente des anomalies. L’accouchement se déroule à Sainte-Justine sous haute supervision médicale. La petite Lyana passe ses deux premières semaines à l’hôpital. Le diagnostic tombe quelques semaines plus tard : syndrome de Noonan, une maladie génétique qui touche environ un enfant sur 2000.
« Généralement, il y a un assez bon taux de survie et les gens touchés ont une assez bonne qualité de vie. Mais il y a une minorité qui est très, très affectée, au point où ça met en danger la vie des nouveau-nés », explique le Dr Andelfinger, qui a suivi la petite dès sa naissance.
Lyana est tombée du mauvais côté des statistiques. Son cœur souffre d’hypertrophie : il épaissit sans cesse, au point de nuire à la circulation sanguine. À la maison, ses parents apprennent à lui injecter dans la bouche, à l’aide d’une seringue, un médicament contre les arythmies cardiaques. La petite est sous haute surveillance médicale, mais elle boit du lait et semble se développer normalement. Ses parents sont encouragés.
Pas de greffe possible
En février 2017, alors que Lyana n’a même pas 3 mois, une équipe médicale du CHU Sainte-Justine convoque cependant les parents.
« Ils nous ont dit que ça ne marchait plus du tout. Que son cœur était tellement hypertrophié qu’il n’y avait rien à faire. »
— Lynn Ménard
La petite est trop jeune pour subir une greffe de cœur. Les petits cœurs de cette taille, de toute façon, sont pratiquement introuvables.
« Elle n’avait jamais montré de signe de faiblesse ou de problèmes. Pour nous, c’est ce qui était le plus difficile. Elle avait l’air d’un bébé normal, elle avait commencé à sourire… », dit son père.
À la maison, les parents passent le plus de temps possible avec leur fille, croyant vivre leurs derniers moments avec elle. Entre ça, ils essaient tant bien que mal de gérer leurs émotions, de s’occuper de leurs autres enfants, de répondre aux questions des proches au meilleur de leurs connaissances.
Dans le salon des Ménard-Deslauriers, il y a un petit écriteau sur lequel on peut lire : « Pour certains, c’est le chaos. Pour nous, c’est la famille. » Ici, le dicton veut vraiment dire quelque chose.
Ce que les parents de Lyana ignorent alors, c’est qu’à Sainte-Justine, un branle-bas de combat s’organise. Dans son bureau du CHU Sainte-Justine, le Dr Gregor Andelfinger montre à La Presse un schéma fait de ronds colorés indiquant des gènes qui sont reliés par des flèches.
Mutations génétiques. Cascade biochimique. Dérèglements. En clair, on comprend que le syndrome de Noonan est déclenché par des mutations sur certains gènes qui poussent une protéine appelée MEK à s’activer. Lorsque cette dernière s’emballe ainsi, elle incite plusieurs cellules du corps à croître et à proliférer de façon incontrôlée. C’est cette prolifération qui explique notamment l’épaississement du muscle cardiaque observé chez plusieurs personnes atteintes du syndrome de Noonan.
Or, il se trouve que la prolifération incontrôlée des cellules est trop bien connue des médecins. Elle explique une autre maladie, beaucoup moins rare que le syndrome de Noonan : le cancer.
« Ce qui est ahurissant dans cette histoire, c’est que la voie de la protéine MEK est aussi la voie qui est concernée dans environ 30 % des cancers chez l’adulte », explique le Dr Andelfinger.
Jamais testé chez un humain
À Sainte-Justine, on réfléchit à toute vitesse. Il existe un médicament anticancer appelé trametinib capable de calmer les ardeurs de la protéine MEK. On l’a même déjà testé sur des poissons et des souris atteints du syndrome de Noonan. Mais jamais la thérapie n’a été essayée chez un être humain pour cette condition, encore moins chez un bébé.
De concert avec plusieurs spécialistes, dont Marie-Ange Delrue, médecin généticienne à Sainte-Justine, le Dr Andelfinger contacte des experts du monde entier. En Europe, les gens de Sainte-Justine tombent sur une équipe aux prises avec un cas similaire – un nouveau-né atteint du syndrome de Noonan en train de dépérir. Administrer du trametinib chez ces enfants très malades comporte des risques qui sont absolument inconnus.
« En mettant tout ce qu’on connaissait ensemble, on s’est dit : il faut essayer, raconte le Dr Andelfinger. On était hors du cadre d’un protocole de recherche. On voulait faire de notre mieux, sur une base compassionnelle. Le point à 1 million de dollars, c’est qu’on n’aurait jamais fait ça sur un enfant qui avait une atteinte viable. »
L’équipe soumet l’idée aux parents de Lyana. « On n’a même pas pris 30 secondes pour y penser, dit la mère de Lyana. C’est sûr que c’était oui. »
Au CHU Sainte-Justine, l’équipe médicale est confrontée à un autre problème : déterminer la dose de trametinib qui a le plus de chances de maximiser les bénéfices en minimisant les risques.
« On n’avait absolument aucune ligne directrice. C’est un travail d’équipe qu’on a fait avec des collègues en génétique, en cardiologie, en pharmacie et en pharmacologie », raconte le Dr Gregor Andelfinger.
Les premiers traitements sont administrés à l’hôpital. Lyana réagit bien. Outre une petite éruption cutanée rapidement calmée avec des crèmes, elle ne présente pas d’effets secondaires.
Dire que la vie devient alors un long fleuve tranquille pour les parents serait toutefois mentir. Chaque semaine, ils doivent se rendre à l’hôpital avec Lyana pour vérifier sa réaction au traitement. À deux reprises, des tests d’imagerie médicale tournent mal. La petite fait d’abord une mauvaise réaction au calmant qu’on lui administre afin qu’elle reste immobile dans l’appareil de résonance magnétique.
« Elle est devenue toute blanche avec les lèvres mauves. Tout le monde s’est mis à courir partout », raconte sa mère. L’affaire finit aux soins intensifs. La deuxième tentative, cette fois sous anesthésie générale, provoque une frousse similaire.
« On a complètement renversé la vapeur »
Une intervention chirurgicale au cœur est planifiée, puis annulée. Épuisés, les deux parents feront tour à tour une dépression. Mais les tests médicaux montrent que le cœur de Lyana s’améliore. Non seulement l’hypertrophie cesse de progresser, mais en plus elle régresse. Le traitement fonctionne, au point où le cœur de la petite, aujourd’hui, est pratiquement normal.
« On a complètement renversé la vapeur », se réjouit le Dr Andelfinger. En Europe, pendant ce temps, l’autre bébé traité réagit de la même façon. Les résultats de cette double expérience sont rapportés aujourd’hui dans la revue Journal of the American College of Cardiology.
L’avenir ? Tant pour la petite Lyana que pour la recherche médicale, il est à la fois prometteur et rempli d’interrogations.
« La grande question à laquelle nous n’avons pas de réponse, c’est : combien de temps il faut la traiter ? Les effets secondaires du traitement sont inconnus, surtout à long terme. »
— Le Dr Gregor Andelfinger
Les médecins tenteront bientôt de sevrer graduellement Lyana de son médicament pour voir comment elle réagit.
En parallèle, ils veulent déployer une véritable étude clinique pour étudier formellement l’effet du trametinib sur les enfants atteints du syndrome de Noonan.
« Il est clair, clair, clair que notre thérapie n’est pas la solution miracle. J’insiste là-dessus », dit le Dr Andelfinger, qui affirme toutefois qu’il s’agit d’une démonstration éloquente de l’importance de la recherche fondamentale.
« Avec les diagnostics génétiques précis d’aujourd’hui et les connaissances des machineries moléculaires, ça devient concevable d’interférer avec le dérangement », dit le Dr Andelfinger.
« Ce traitement a tout changé, dit quant à lui Joël Deslauriers, le père de Lyana. Tu passes d’un moment où tu crois que ta fille va mourir à une vie de famille avec tous ses membres. Ça vient avec des embûches et beaucoup d’incertitude, mais elle est avec nous aujourd’hui et pour très longtemps. Lyly est une battante. Elle va passer à travers tout. »