


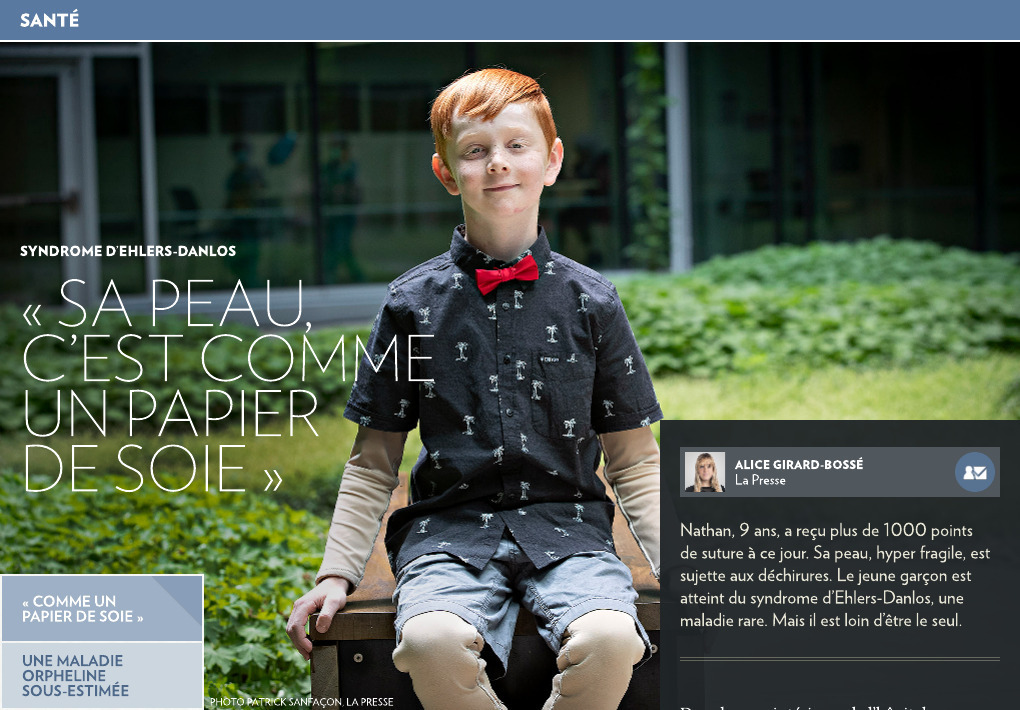
« Sa peau, c’est comme un papier de soie »
Dans la cour intérieure de l’hôpital Sainte-Justine, Nathan joue avec son chiot Mira en compagnie de sa grande sœur Chloé et de son petit frère Gabriel. Outre quelques cicatrices sur son front, rien ne pourrait laisser imaginer que le jeune garçon de 9 ans a la peau aussi fragile que du papier de soie.
« Quand Nathan est né, on l’appelait le bébé Jell-O. On aurait dit qu’il n’avait pas de tonus, il pliait, on trouvait ça un peu drôle », raconte sa mère, Julie Desroches. Quand le bambin a commencé à se déplacer par lui-même et qu’il se cognait, il développait immédiatement des ecchymoses. « Quand les bleus ont commencé à fendre, on s’est posé beaucoup de questions », se remémore-t-elle.
La famille fait évaluer Nathan en hématologie. Rien d’anormal n’est détecté. À 18 mois, un ami de la famille, médecin, leur recommande alors d’aller passer le test pour le syndrome d’Ehlers-Danlos. Le test est positif. Il est bien atteint de cette maladie génétique.
Le syndrome d’Ehlers-Danlos est une maladie rare qui touche la production de collagène. Cette protéine est essentielle pour la cicatrisation des plaies, pour l’élasticité de la peau et pour maintenir les tissus ensemble. Environ 1 personne sur 5000 est atteinte de la maladie.
« Sa peau, c’est comme un papier de soie, dès qu’il l’accroche, elle se déchire. Normalement, quand on se coupe, ça fait une belle ligne droite, mais Nathan, sa peau se déchire n’importe comment et ça fait des plaies très larges. »
— Julie Desroches, mère de Nathan
Aucun suivi
Après le diagnostic, aucun soutien n’a été offert à la famille. Les parents ont été laissés à eux-mêmes. « Avec les maladies rares, c’est dur d’avoir de l’aide. Les parents doivent chercher beaucoup. Il n’y a pas de suivi. Tu reçois un diagnostic et tu ne sais plus quoi faire après », confie Mme Desroches.
Après de nombreuses recherches, la famille contacte l'Hôpital juif de réadaptation, à Laval, pour que Nathan y soit suivi. « Il était jeune et il était petit, alors on s’est fait refuser, parce qu’il n’entrait pas dans les critères », se remémore sa mère.
Nathan a reçu son diagnostic à 18 mois, ce qui est une exception. « C’est très jeune. Il y en a qui le découvrent dans la trentaine », dit-elle. Le jeune garçon a donc été transféré à l’hôpital Marie-Enfant, puis au CHU Sainte-Justine.
1000 points de suture
Le Dr Daniel Borsuk, chef de la chirurgie plastique au CHU Sainte-Justine et connu pour avoir effectué la première greffe de visage au Canada, a pris Nathan sous son aile. Grâce à son équipe, il a développé une technique spéciale pour réparer les blessures du garçon.
Chez les enfants, le fil utilisé pour faire les points de suture est fondant au contact de la peau et disparaît en une à deux semaines. Le problème ? Nathan a une durée de cicatrisation beaucoup plus longue allant de six à huit semaines.
« En une à deux semaines, Nathan n’est pas guéri. Alors si tu mets des points fondants, toute la plaie va rouvrir. Même si la plaie est minime, il faut mettre des points permanents », explique le Dr Borsuk. Pour permettre une meilleure guérison, le plasticien referme sa peau en plusieurs niveaux.
Nathan a reçu, jusqu’à présent, plus de 1000 points de suture. Le jeune garçon est devenu un expert pour se les faire poser. Pendant les interventions, il chante des chansons, il joue à des jeux avec sa famille et il discute avec les médecins.
Le plasticien se souviendra toujours de sa première rencontre avec son jeune patient.
« Il n’a pas bougé et il m’a regardé faire les points. Il n’avait même pas 4 ans et il était déjà habitué d’avoir des points de suture. Normalement, les enfants crient et pleurent. »
— Le Dr Daniel Borsuk, chef de la chirurgie plastique au CHU Sainte-Justine
Moins de blessures
Dans les premières années de sa vie, Nathan se retrouvait souvent à l’hôpital. « Il se blessait toutes les deux semaines », dit sa mère. Afin de limiter les blessures, sa famille a dû réorganiser la maison en installant des tapis de mousse dans plusieurs pièces.
À 9 ans, Nathan est maintenant plus conscient des risques de blessures et il porte des vêtements de protection en tout temps. Ce sont des vêtements coussinés faits sur mesure, que la famille renouvelle chaque année.
« Pour quelqu’un qui le voit, il a juste l’air de porter un chandail en dessous de son t-shirt. Il vit très bien avec ça, car il est très conscient que ses protections, c’est un peu garant de son plaisir, de son bonheur et de sa vie », affirme Mme Desroches.
Les assurances ne remboursent toutefois pas ces vêtements très coûteux. « Comme c’est un vêtement de prévention, les assurances ne les couvrent pas. Chaque année, on cherche des subventions », dit-elle.
Des centaines de suivis
La maladie de Nathan touche la production de collagène dans tous les tissus conjonctifs de son corps. Outre la fragilité de sa peau, son syndrome touche également ses tendons, ses ligaments et ses organes. Tout ce qui est composé de tissus conjonctifs peut se dégrader.
« Nathan est suivi en cardiologie, parce qu’il a un souffle au cœur, en orthopédie parce qu’il a une scoliose, en orthophonie à cause de l’hyperlaxité au niveau de sa mâchoire, en ergothérapie parce que ses doigts se plient à l’envers quand il prend des objets et en ophtalmologie parce que ses yeux peuvent se dégrader », énumère sa mère. C’est sans compter ses suivis multiples en génétique, en dermatologie et en plastie.
Tous ces spécialistes rencontrent le jeune garçon tous les six mois ou chaque année. « Si on regarde dans son dossier, il a eu des centaines et des centaines de rendez-vous à l’hôpital », dit le Dr Borsuk.
Jouer comme un enfant
Le syndrome n’empêche pas Nathan d’avoir une vie normale. Il est dans une école ordinaire et n’a pas de retard scolaire. Une préposée l’accompagne 25 heures par semaine. « Elle veille sur lui lors des déplacements, elle fait les adaptations lors des cours d’éducation physique et elle est formée en premiers soins », indique la mère.
En dehors des classes, ses parents, deux professeurs d’éducation physique, l’encouragent à bouger et à s’épanouir comme les autres enfants. « Il est excellent en sports », dit sa mère. Il fait du patin, mais ne joue pas au hockey pour éviter les contacts. Il a également été en ski pour la première fois à l’hiver 2020. « J’aime beaucoup le golf. J’ai joué deux fois cette année », lance Nathan avec un sourire.
« C’est un enfant, alors il joue comme un enfant, renchérit le Dr Borsuk. Il a des cicatrices au visage et il s’en fout, il se trouve beau. Il a une joie de vivre incroyable », conclut-il.